
在《科学进展》杂志封面上刊登的一篇新报告中,Hans Hon Sang Chan和牛津大学材料、化学和量子光子学研究团队生成了具有多达36个量子位的精确模拟量子计算机,以探索资源节约算法,并模拟具有单个和成对粒子的2D 和 3D原子。
化学建模是量子计算机的一个自然属性,尽管现有方法无法开发近乎完美的量子比特。在这项工作中,量子化学家探索了从基态制备和能量估计到电子散射和电离动力学的一系列任务,以评估分裂操作模拟中的各种方法,以便模拟一些感兴趣的分子的量子化学。基于网格的方法表现出色,为不易出错的量子计算时代让路。
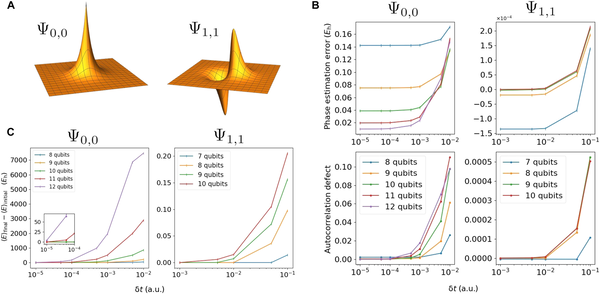

二维氢电子的SO-QFT模拟。(A)二维氢的基态ψ0,0(左)和第一次激发态ψ1,1(右)的实际投影。请注意,此处的绘图不反映模拟框大小的选择,并且不按比例缩放。(B) 顶部为相位估计能量与二维氢解析能量之差。底部捕获传播结束时模拟保真度的偏差。在这一系列实验中,我们初始化了以L = 10 a.u为中心的基态ψ0,0,使得库仑奇点的起源位于两个中心网格点之间。每个子寄存器的预算为 0 ≤ nr ≤ 0 个量子位来存储波函数,对应于 10.8 ≥ δr ≥ 12.0 a.u. 的空间分辨率。我们还初始化ψ1,1激发态40 a.u模拟盒子的长度,与预算7≤nr≤10量子位/ subregister和相应的决议0.313≥δr≥0.039 a.u。这两种状态以不同的空间分辨率表示,都使用一阶time-propagated 1.5原子时间单位。对于ψ0,0状态,我们使用时间步长在0.0001≤δt≤0.01之间(15万到150个SO步长),对于ψ1,1状态,我们使用时间步长在0.001≤δt≤0.1之间(1500到15个SO步长)。(C)最终和初始能量期望值之间的差异,通过对状态的直接采样测量,基态(左)和第一激发态(右),以不同的空间和时间分辨率传播。左边的插图放大了基态在高时间分辨率下的能量误差。
通过实空间网格方法进行量子计算
量子化学家将量子计算机设想为化学预测和探索的变革性工具。虽然传统计算机可用于探索量子分子动力学以预测反应结果和实验观察结果,但硬件成本和持续时间可能会随着模拟粒子的数量呈指数增长。在这项工作中,Chan 及其同事研究了基于真实空间网格方法在早期版本的量子计算机上加速化学动力学模拟的基本特征。
这些早期版本的量子计算机具有有限数量的纠错量子比特。该团队对粒子对称性等特征进行编码,以在研究过程中为复杂有趣的分子提供最佳的资源缩放。
大多数量子计算机都有噪声,而且成本高昂。因此,研究人员采取了一种不同的方法,通过部署经典的计算资源来模拟小型但无噪声的量子计算机,从而模拟其中的量子分子动力学,从而直接检查成本和性能指标。虽然他们没有重新使用现有的经典网格技术来进行基于网格的模拟,但他们为与化学相关的量子动力学进行了真实的、无噪声的量子机器的模拟。
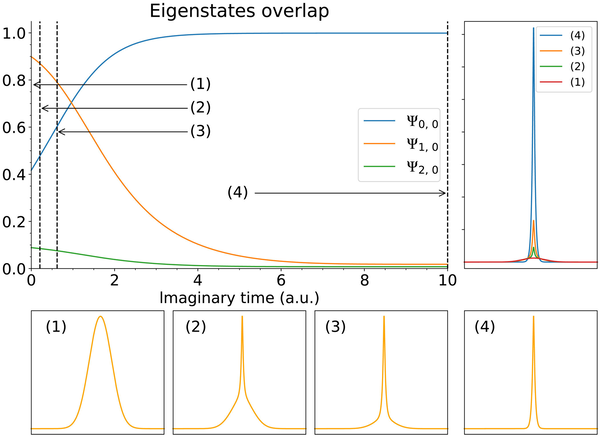
使用 PITE 技术制备二维氢的基态。该方法在 1 + 2 × 10-量子比特量子计算机上进行了模拟。始终选择成功的结果。主图显示传播态与解析本征态的重叠。底部显示了在虚时间演化期间在标记点采样的电子概率密度的缩放横截面。右上角显示了以相同比例绘制的相同概率密度。
量子化学的新方法
模拟的成本将量子计算机模拟限制在包含 36 个完美量子位的中等尺寸版本。该团队使用实验装置探索了几个信息场景,用于单电子和双电子系统的 2D 和 3D 模拟。他们选择了化学中两个关键的兴趣领域,并估计了模拟强外场动力学所需的量子资源,然后模拟了粒子散射动力学。
在第一个实验中,该团队突然施加了一个外场,导致偶极子振荡和单个束缚电子的电离。他们设想在这个方向上的努力将包括光化学和激光激发等主题。物理学家和量子化学家认为小分子的相干量子调控是化学科学的“圣杯”之一。例如,该过程可以让科学家在氢原子去除的背景下研究氨,以探索其在现代农业中的潜力。
在第二种情况下,该团队检查了与光谱学、天体化学和制造过程相关的电子分子散射,因为碰撞和散射过程是高度动态的,因此很难进行经典建模。
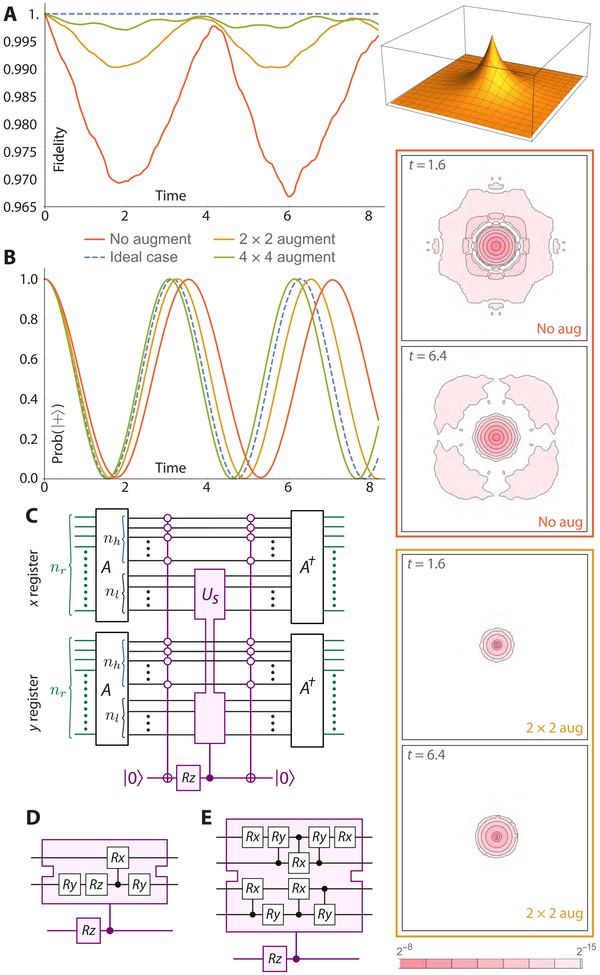
ASO 技术的性能。模拟的量子计算机有 13 = 1 + 6 × 6 个量子比特。(A) 3D 插图描绘了其模拟盒内 2D 氢的本征态 ψ0,0。该图显示了时间 t 的状态相对于初始状态的自相关,理想情况下初始状态保持一致(蓝色虚线)。红色、橙色和绿色线分别是无增强、2×2 增强和 4×4 增强的情况。(B) 相同三种情况的相位估计结果,理想情况再次用蓝色虚线显示。右边的等高线图显示了概率密度的绝对差异,相对于初始状态,在没有增强(红色)和 2 × 2 增强(橙色)的情况下。(C) 使用的通用电路。(D) 和 (E) 分别指定了用于我们的 2 × 2 和 4 × 4 增强的特定电路。A 运算符只是增加空间“像素”的索引,以便右下角的像素为 (0,0)。在 2 × 2 增强的情况下,使用的增量为 G = 1,以便 A 映射感兴趣像素的索引,即 (−1,−1)、(−1,0)、(0,− 1), 和 (0,0) 到 (0,0), (0,1), (1,0), 和 (1,1), 分别。这些索引现在正是那些最重要的 nr − 1 量子位为零的状态,便于将小型增强电路仅应用于四个目标状态。信用:使用的增量为 G = 1,因此 A 将感兴趣像素的索引 (−1,−1)、(−1,0)、(0,−1) 和 (0,0) 分别映射到 (0,0)、(0,1)、(1,0) 和 (1,1)。这些索引现在正是那些最重要的 nr − 1 量子位为零的状态,便于将小型增强电路仅应用于四个目标状态。
波浪模拟
科学家们使用分裂算子量子傅里叶变换(SO-QFT)哈密顿模拟方法来执行波包操作,并提出了一系列结果,这些结果适用于使用单个和成对粒子的 2 到 3D 系统的基于网格的方法。根据结果,他们评估了模拟所需的量子资源,以提供合适的量子硬件架构。
数值结果导致了通过QuEST、QuEST-link和pyQuEST等开源工具实现量子处理器的模拟。他们探索了量子位的数量,并估计了执行的持续时间,以实现给定精度的模拟,并研究了一种基于采样的方法来估计系统能量的场景,该方法被证明对缺陷高度敏感。他们估计了随之而来的量子资源成本,并指出了合适量子计算机的硬件布局。
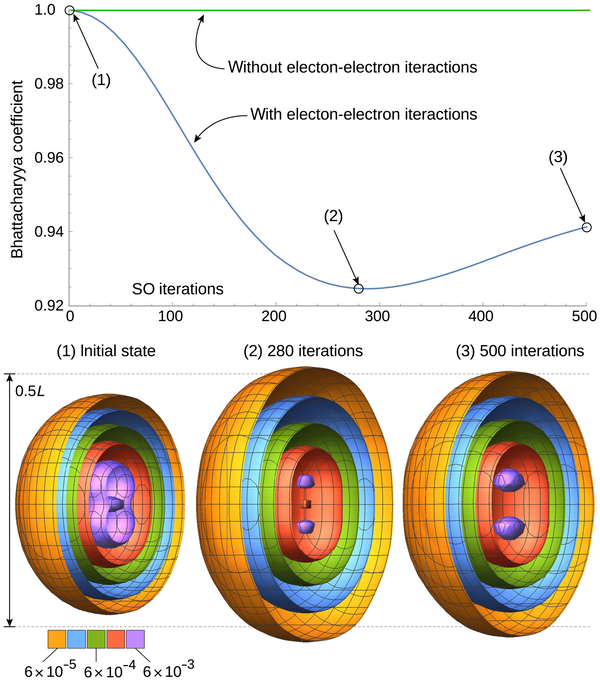
真实空间中氦原子的模拟。上图是 Bhattacharyya 系数(启用和不启用电子-电子相互作用),下图是氦原子模拟时间演化过程中的真实空间电子密度分布。彩色壳是电子概率密度等值面,在 L = 25 au 的模拟框中显示了初始状态、最大展开时间和模拟结束时的分布。500 个 SO 周期对应于 25 au (≈0.6 fs) 的传播。
方法和 3D 模拟
研究小组探索了单辅助迭代相位估计(IPE)测量来预测激发态。IPE电路在近期的量子计算实践中是有趣和重要的。研究结果用于能量估计,该团队在基于概率虚时间演化(PITE)的量子计算机上进行了制备真实空间基态的替代方法,并模拟了2D氢的基态,同时详细说明了该方法的缺点。他们进行了两种量子动力学模拟,这两种模拟依赖于两种情况:(1)由强外部场电离;(2)依赖于电子-电子散射。
接下来,该团队引入了增强分裂算子(ASO),通过向基本SO-QFT(分裂算子量子傅里叶变换)周期提供额外的元素来优化模拟的保真度。如本文所述,在严格库仑相互作用下进行所有数值研究时,ASO方法是高度相关的。利用这个装置,他们在3D中模拟了氦原子的动力学。他们使用薛定谔方程来近似氦原子的真实电子本征态,并使用巴塔查里亚系数来表示氦原子模拟的电子-电子相互作用和时间演化。
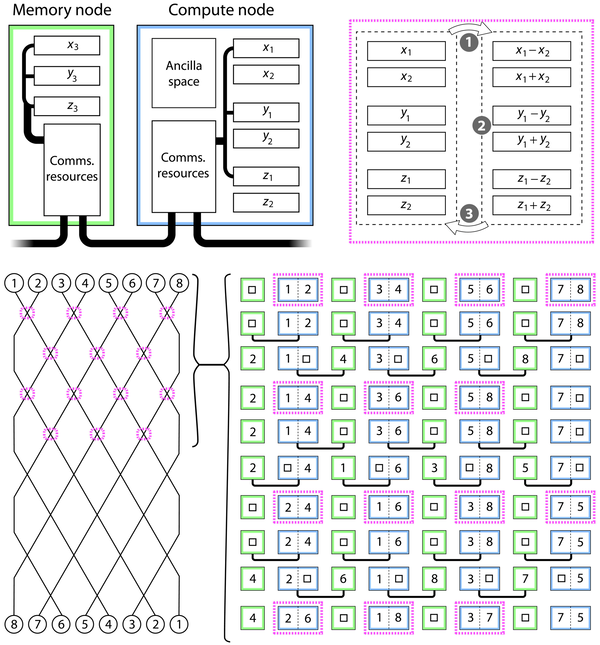
一种可能的“多核”架构。基于将 P 粒子状态分布在 P/2 个存储节点(3nr 量子位)和 P/2 个计算节点(6nr 量子位)上,这些节点在芯片上或宏观尺度上相互关联。右上表示三个步骤发生在计算节点中。在步骤 1 中,使用量子加减法移动到相对和总坐标(步骤 3 将反转此过程)。在中间的步骤 2 中,相对坐标用于应用 SO 循环所需的相移,并且可选地,我们应用增强步骤。底部显示了并行化阶段如何交替处理和排列数据。圆圈和方块内的数字是粒子的标签;它们对应于右上角的 x、y 和 z 符号的下标。
量子计算资源和架构
量子化学家研究了进行超出经典算法范围的量子建模所需的资源,并调整了适合此类表达式的量子架构。他们估计了六氟乙烷(C2F6)和氨(NH3)分子模拟感兴趣的量子场景所需的量子位的数量。基于网格的C2F6模拟需要大约2250个计算量子比特,而氨分子只需要不到450个量子比特。
模拟的时间成本还取决于硬件实现。因此,相对于深度算法和错误率,最容易理解的代码需要每个逻辑量子比特有数百个物理量子比特,这与当今最好的量子计算机原型相当。研究人员还设计了一种多核网络架构来支持理论上的大规模量子比特。
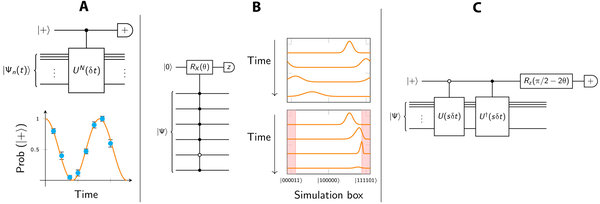
在这项工作中探索了三种用于真实空间化学的早期容错量子电路技术。(A) 本工作中模拟的单辅助 IPE 方法。全局相位以测量纠缠的辅助量子比特的概率进行编码,该量子比特控制在︱+⟩状态下的N SO周期的应用。为了获得这一概率,必须在希望对信号进行采样的每个时间点重复传播和测量。(B) 量子位寄存器上波包的衰减。右上角描绘了高斯波包在周期性边界上的色散。添加粉红色的复合吸收区域(右下)通过降低其范数来衰减散射波包。在图示的案例中,这个过程并不十分完美:衰减过于严重会导致一些反射。左边是在选定像素处执行概率波包衰减的电路。选中像素︱111101⟩,对应图中模拟框右侧的衰减区域。(C) 使用 PITE 电路准备基态。实心圆圈表示“由 |1⟩ 控制”,空心圆圈表示“由 0 控制”。在∣+⟩结果上进行后选择会产生一个状态,其中一阶应用了虚时间演化步骤。
展望
通过这种方式,Hans Hon Sang Chan 及其同事探索了分裂算子量子傅立叶变换 (SO-QFT) 方法来模拟精确的量子位并测试真实空间量子化学模拟背后的技术。他们探索了几种已知的量子技术,并介绍了其他一些技术来传达量子模拟的关键方面。科学家们描述了在早期容错量子计算机上实现数字实验的基础资源。
结果可以是一个学习/预测周期,通过机器学习增强物理实验来加速化学发现。这些结果可以引导量子技术的各个领域,包括用狭义相对论来模拟高能粒子,以及在金融工程中发挥作用。
参考链接:
https://phys.org/news/2023-03-quantum-chemistry-simulations.html
https://www.science.org/doi/10.1126/sciadv.abo7484
—煤油灯科技victorlamp.com编译整理—
版权声明:本文内容转自互联网,本文观点仅代表作者本人。本站仅提供信息存储空间服务,所有权归原作者所有。如发现本站有涉嫌抄袭侵权/违法违规的内容, 请发送邮件至1393616908@qq.com 举报,一经查实,本站将立刻删除。